非洲裔人群中,APOL1基因的高风险变体(G1、G2)与肾小球疾病的高发密切相关,但其致病机制长期未明。今年10月,来自荷兰莱顿大学医学中心(LUMC)与美国维克森林大学医学院的联合研究团队,通过患者来源诱导多能干细胞(iPSC)构建的肾类器官模型,系统解析了APOL1变体如何在足细胞中诱导代谢重编程与线粒体功能障碍。
研究显示,APOL1风险等位基因可在炎症刺激下触发能量代谢失衡、氧化磷酸化下降与糖酵解增强,最终导致足细胞应激与功能衰退。该工作为阐明APOL1相关肾病的分子机制提供了关键证据,也为代谢干预策略打开了新方向。
该研究提出了一个新的病理框架:APOL1风险变体通过抑制线粒体代谢与能量信号,引发足细胞代谢重编程与功能衰竭。这一模型连接了遗传变异、能量稳态失衡与肾小球损伤三者之间的因果链条。未来工作可进一步探索:利用空间转录组与代谢组揭示APOL1病变在肾小球微环境中的传播模式、通过代谢激活剂或AMPK通路调节剂验证临床可行性、以及构建多模态“APOL1-代谢-免疫”系统模型,为高风险人群早期筛查与干预提供新思路。
研究亮点
1. 患者来源+等基因对照的人源模型:用两名G1/G1、G2/G2纯合患者的iPSC构建肾类器官,并在G2背景上通过CRISPR-Cas9生成等基因G0对照;在IFN-γ刺激后,APOL1在足细胞层面转录与蛋白均上调。
2. 足细胞是主要受影响细胞群:APOL1与PODXL⁺足细胞共定位;在足细胞谱系中,晚期足细胞表达最高,且其失调基因数量在所有肾单位细胞中最多。
3. 代谢重编程(由OXPHOS转向糖酵解):风险变体(RV)晚期足细胞表现为OXPHOS/线粒体呼吸通路下调,而糖酵解与HIF-1缺氧信号上调。
4. RV特异的“糖酵解晚期足细胞”亚群:G1/G2中可见、在G0中几乎缺失;该亚群富集SLC2A1、ENO1、LDHA、BNIP3等糖酵解/缺氧标志,并伴随胶原合成/修饰相关基因升高。
5. 功能与形态学证据指向线粒体受损:同位素示踪与呼吸实验显示糖酵解通量增强、TCA活性降低;G0肾小球在IFN-γ后最大呼吸上升,而G1/G2反应被钝化;iPSC衍生足细胞出现线粒体分支数与分支长度下降的碎裂化表型。
6. 空间脂质组学用于精确定位成熟足细胞:MALDI-MSI+免疫染色整合识别13个脂质簇,并以PODXL⁺/NEP⁺标记成熟足细胞,为在其上开展动态代谢与功能测定提供定位基础。
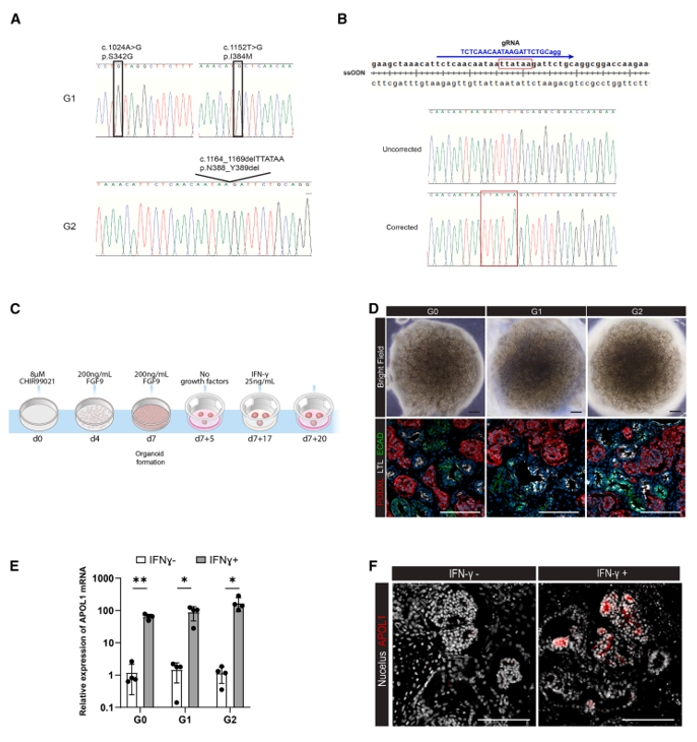
图1. 研究团队构建APOL1相关肾病(AMKD)患者来源及等基因对照iPSC肾类器官模型的全过程。研究首先对携带G1G1与G2G2风险变体的患者iPSC进行测序验证,并利用CRISPR-Cas9基因编辑将G2变体修复为等基因G0G0对照系(红框标示突变缺失位点,gRNA与PAM序列用于编辑定位)。随后,三种细胞系(G1、G2、G0)均经标准诱导分化生成肾类器官,并在干扰素γ(IFN-γ)刺激后3天收集进行分析。显微与免疫染色显示,所得类器官成功形成包含足细胞(PODXL⁺)、近端小管(LTL⁺)与远端小管(ECAD⁺)的结构。定量PCR结果表明,APOL1 mRNA在IFN-γ处理后显著上调(p<0.05),免疫荧光进一步证实了蛋白水平的APOL1表达增强,为后续解析变体特异性代谢效应奠定基础。
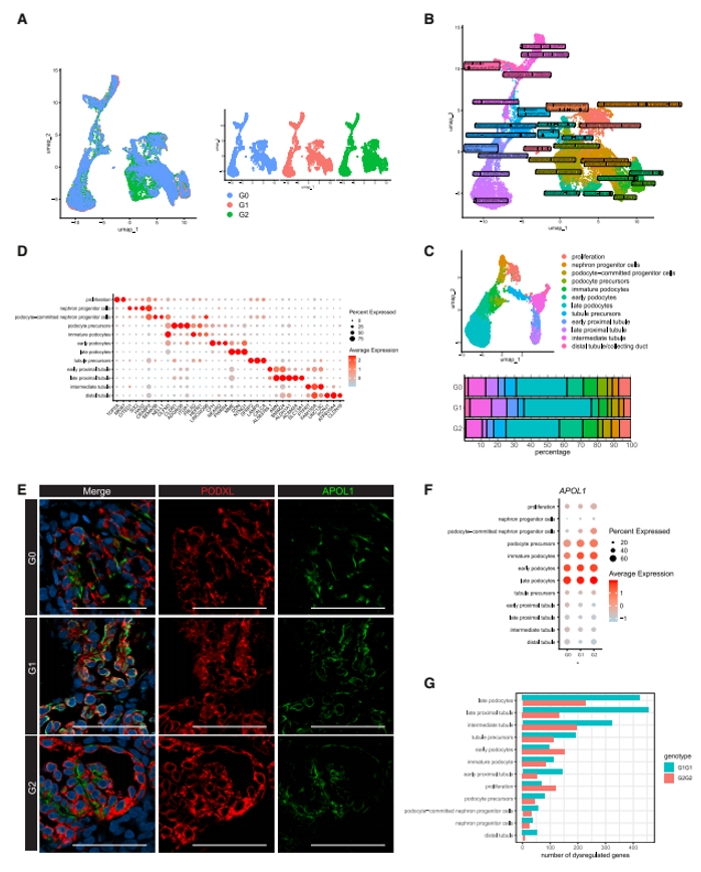
图2. 揭示了APOL1在肾类器官中主要定位于足细胞,并显示风险变体(RV)足细胞的转录失调最为显著。UMAP降维分析显示,从G0、G1和G2三种iPSC来源的肾类器官中共识别出约30个细胞群体,涵盖肾单位主要细胞类型,各基因型间肾单位细胞组成比例相似。标志基因表达与免疫染色结果一致,APOL1主要与PODXL⁺足细胞共定位,在足细胞前体、未成熟与成熟阶段均有表达,而其他细胞类型中表达极低。差异分析进一步显示,风险变体足细胞(G1/G2)拥有最多的转录失调基因,提示APOL1变体特异性地引发足细胞的广泛基因表达重编程,为其致病机制提供分子依据。
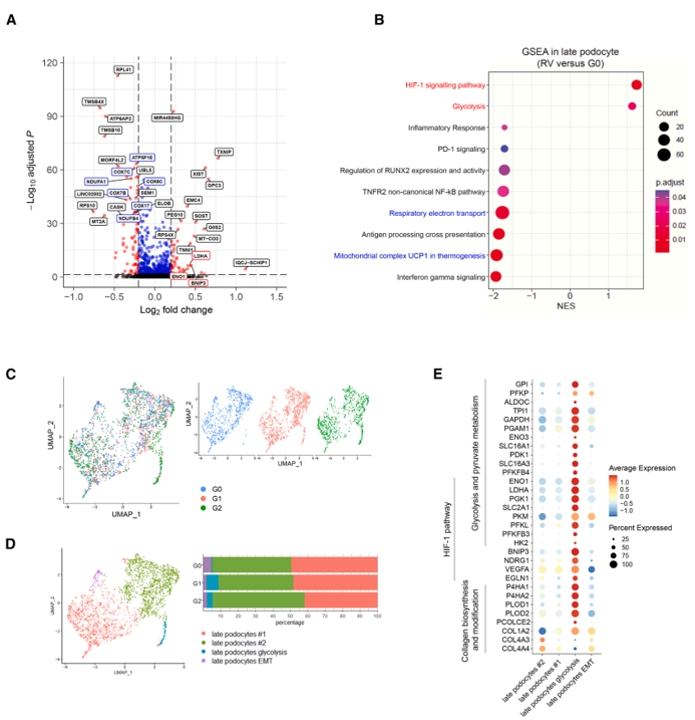
图3. APOL1风险变体在晚期足细胞中触发代谢通路的系统性重编程:差异表达与基因集层面的结果共同指向氧化磷酸化(OXPHOS)下调、糖酵解与HIF-1缺氧信号上调,呈现由OXPHOS向糖酵解迁移的趋势。进一步的晚期足细胞亚群分析发现,一个以糖酵解为特征的亚群在G1/G2中可见、而在G0中几乎缺失,提示该表型与风险变体特异相关。其标志基因包括SLC2A1、ENO1、LDHA、BNIP3,并伴随胶原生物合成/修饰基因(如P4HA1/2、PLOD1/2、COL1A2)特征增强,暗示能量代谢改变与基质重塑可能并行发生。
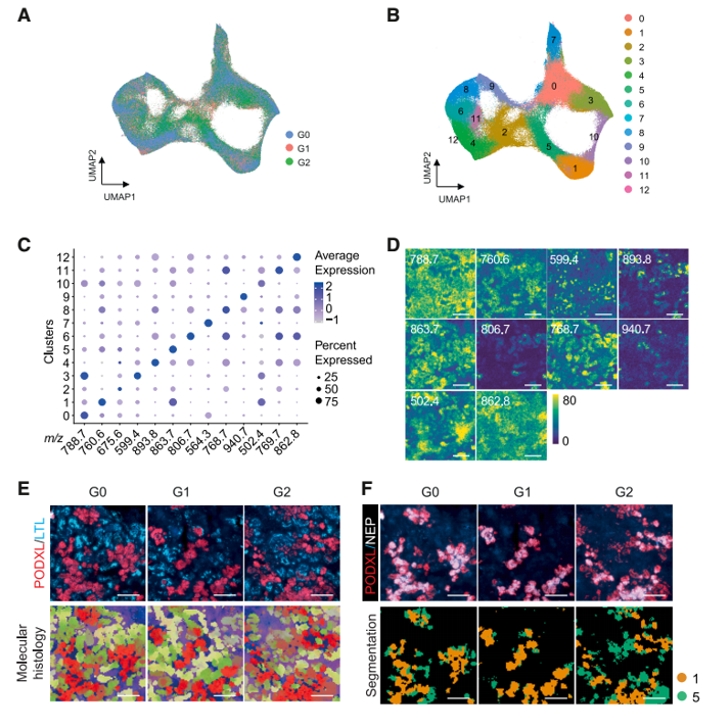
图4. 利用空间脂质组学(MALDI-MSI)+免疫染色联用,在肾类器官中实现对成熟足细胞的定位与分型:对G0/G1/G2在7+20天的类器官进行脂质组数据整合可见三种基因型聚类相近,并基于脂质特征识别出13个簇并回映到组织切片上;随后将PODXL/LTL免疫信号与MSI结果配准,生成像素级的“分子组织学”可视化。进一步以PODXL与NEP(MME)双阳性界定成熟足细胞,发现脂质簇1(特征m/z 760.6)与PODXL⁺/NEP⁺区域重叠,而脂质簇5对应PODXL⁺/NEP⁻的未成熟足细胞,提示成熟/未成熟足细胞存在可分辨的空间脂质异质性。
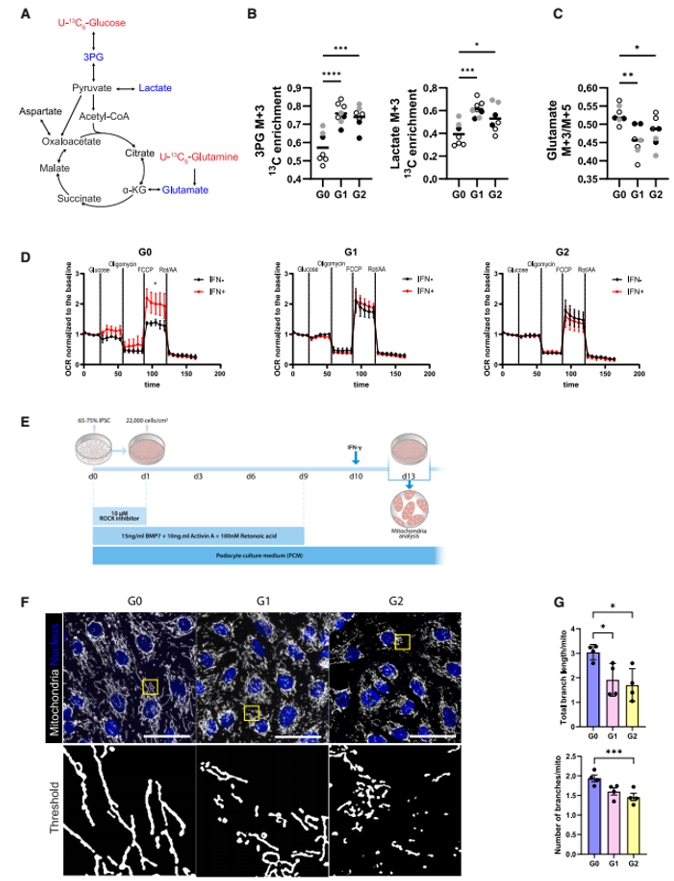
图5. 空间同位素示踪+呼吸功能与形态学多证据表明:APOL1风险变体(RV)使成熟足细胞发生代谢与线粒体损伤。具体而言,U-¹³C₆-葡萄糖示踪显示3PG与乳酸(M+3)标记显著升高,提示糖酵解通量增强;U-¹³C₆-谷氨酰胺示踪显示谷氨酸 M+3/M+5 比值下降,指向TCA循环活性降低。呼吸应激实验中,G0肾小球在IFN-γ后最大呼吸上升,而G1/G2反应被显著钝化,提示电子传递链适应性受损。在2D iPSC来源足细胞中,IFN-γ诱导3天后可见线粒体分支数与总分支长度显著下降,呈碎裂化,进一步支持线粒体功能障碍是RV足细胞的核心表型。
意义与展望
该研究提出了一个新的病理框架:APOL1风险变体通过抑制线粒体代谢与能量信号,引发足细胞代谢重编程与功能衰竭。这一模型连接了遗传变异、能量稳态失衡与肾小球损伤三者之间的因果链条。未来工作可进一步探索:利用空间转录组与代谢组揭示APOL1病变在肾小球微环境中的传播模式、通过代谢激活剂或AMPK通路调节剂验证临床可行性、以及构建多模态“APOL1-代谢-免疫”系统模型,为高风险人群早期筛查与干预提供新思路。